St. Petersburg State Technological Institute
(Teknisk universitet)
Institutt for organisk kjemi fakultet 4
Gruppe 476.
Kursarbeid
Oksidasjon av alkener
Student ............................................. Rytin A.I.
Foreleser .................................... ... St. Petersburg Yu.l.
St. Petersburg
Introduksjon
1.Poxidering (reaksjon N.A. PRILEZHEV, 1909)
2. Hydroksilisering
2.1anti. -Hhydroksylering
2.2syn -Hhydroksylering
3. OVISKERING AV ALKENES
4.OSomolyse
5. Samfunn av alkener i nærvær av palladiumsalter
Konklusjon
Liste over kilder brukt
Introduksjon
Oksidasjon er en av de viktigste og vanlige transformasjonene av organiske forbindelser.
Under oksidasjon i organisk kjemi, fører prosessene som fører til uttømming av forbindelser med hydrogen eller anrikning med oksygen. I dette tilfellet finner elektronmolekylet sted. Følgelig forstår restaureringen separasjonen fra det organiske oksygenmolekylet eller hydrogenvedlegget til det.
I oksidative reduksjonsreaksjoner er oksidasjonsmidler forbindelser med en stor affinitet for en elektron (elektrofila) og reduksjonsmidler - forbindelser som har en tendens til å returnere elektroner (nukleofiler). Den enkle oksydasjon av forbindelsen øker med veksten av sin nukleofilisitet.
Når oksiderende organiske forbindelser, som regel, den totale overføringen av elektroner og følgelig, forekommer ikke endringer i valenset av karbonatomer. Derfor er konseptet av oksidasjonsgraden den betingede ladningen av et atom i molekylet, beregnet, basert på antagelsen om at molekylet bare består av ioner - er bare betinget, formell karakter.
Ved fremstilling av ligningene av redoksreaksjoner er det nødvendig å bestemme reduksjonsmidlet, oksydasjonsmidlet og antall hengivne og mottatte elektroner. Som regel er koeffisientene valgt ved hjelp av elektron-ionbalansemetoden (halvformasjonsmetode).
I denne metoden, overgangen av elektroner fra en atomer eller ioner til en annen, med hensyn til naturen til mediet (syre, alkalisk eller nøytral), hvor reaksjonen fortsetter. Å utjevne antall oksygen- og hydrogenatomer eller vannmolekyler og protoner (hvis surmedium), eller vannmolekyler og hydroksydioner (hvis alkalisk) innføres.
Ved å skrive semitraksjoner med utvinning og oksidasjon, er det derfor nødvendig å fortsette fra sammensetningen av ioner som er i oppløsning. Malodikasjoner, dårlig oppløselig eller gass utgitt i form av gass skal skrives i molekylær form.
For eksempel vurdere prosessen med etylenoksydasjon ved en fortynnet vandig oppløsning av kaliumpermanganat (Wagner-reaksjon). I løpet av denne reaksjonen oksyderes etylen til etylenglykol, og kaliumpermanganat blir gjenopprettet til mangansioksid. Ved den dobbelte forbindelsen er to hydroksyl sammenføyet:
3c 2 h 4 + 2kmno 4 + 4h 2 o → 3c 2 h 6 o 2 + 2mno 2 + 2koh
Semi-reaksjonsgjenoppretting: Mno 4 ¯ + 2h 2 o + 3 e. → mno 2 + 4oh ¯ 2Oksidasjon Semi-reaksjon: C 2 H 4 + 2OH - - 2 e. → C 2 H 6 O 2 3
Endelig har vi i ionform:
2mno 4 ¯ + 4h 2 o + 3c 2 h 4 + 6oh ¯ → 2mno 2 + 8oh ¯ + 3c 2 h 6 o 2
Etter å ha utført de nødvendige kutt av slike medlemmer, skriv ligningen i molekylær form:
3C 2 H 4 + 2KMNO 4 + 4 H20 \u003d 3C2H6O2 + 2MNO 2 + 2KOH.
Karakteristisk for noen oksidasjonsmidler
Oksygen
Air oksygen er mye brukt i teknologiske prosesser, da det er den billigste oksidanten. Men oksygenering av luft oksygen er forbundet med vanskelighetene forbundet med kontrollen av prosessen, som strømmer i forskjellige retninger. Oksidasjon utføres vanligvis ved høye temperaturer i nærvær av katalysatorer.
Ozon
Ozon O 3 brukes til å oppnå aldehyder og ketoner hvis de er vanskelige å oppnå på andre måter. Oftest er ozon brukt til å etablere strukturen av umettede forbindelser. Ozon er oppnådd under virkningen av en stille elektrisk utslipp på oksygen. En av de betydelige fordelene med ozonisering, sammenlignet med klorering, er fraværet av toksiner etter behandling.
Kaliumpermanganat
Permanganatkalium er det mest brukte oksiderende middel. Reagenset er oppløselig i vann (6,0% ved 20 ° C), så vel som i metanol, aceton og eddiksyre. For oksidasjon, vandig (noen ganger aceton) løsninger KMNO 4 i et nøytralt, syre eller alkalisk medium. Ved gjennomføring av en prosess i et nøytralt medium, blir magnesium, aluminium eller karbondioksid tilsatt til nøytraliseringen av kaliumfremklaget hydroksyd, tilsettes til reaksjonsmassen. Oksidasjonsreaksjonen av KMNO 4 i et surt medium utføres oftest i nærvær av svovelsyre. Det alkaliske medium i oksidasjon skaper KOH generert under reaksjonen, eller den tilsettes i utgangspunktet til reaksjonsmassen. I svakt alkaliske og nøytrale medier oksiderer KMNO 4 ved ligning:
KMNO 4 +. 3 e. + 2h 2 o \u003d k + + mno 2 + 4oh ¯
i et surt miljø:
KMNO 4 +. 5 e. + 8h + \u003d k + + mn 2+ + 4h 2 o
Kaliumpermanganat brukes til å oppnå 1,2-dioler fra alkener, med oksydasjon av primære alkoholer, aldehyder og alkylarens til karboksylsyrer, så vel som for oksidativt splitting av karbonskjelettet over flere bindinger.
I praksis brukes ganske stort overskudd (mer enn 100%) KMNO 4 vanligvis. Dette forklares av det faktum at Kmno 4 delvis dekomponerer under normale forhold, dekomponerer KMNO 4 delvis på mangansioksid med utgivelsen av O 2. Dekomponert konsentrert H2SO4 ved oppvarming i nærvær av gjenoppbyggere med en eksplosjon; Blandinger av kaliumpermanganat med organiske stoffer er også eksplosiver.
Nadkislot.
Peroteletisk og permaraye syrer oppnås ved en reaksjon på 25-90% hydrogenperoksid med en passende karboksylsyre i henhold til følgende reaksjon:
RCOOH + H20 2 \u003d RCOOOH + H 2 O
I tilfelle av eddiksyre settes denne likevekten relativt sakte, og for å akselerere dannelsen av topper tilsettes vanligvis svovelsyre som en katalysator. Myresyren er sterk nok i seg selv for å sikre rask etablering av likevekt.
Penrophunturuce syre oppnådd i en blanding med trifluoreddiksyre-reaksjon av trifluoreddiksyreanhydrid med 90% hydrogenperoksid, en enda sterkere oksidasjonsmiddel. På samme måte kan Perussesyre oppnås fra eddiksyreanhydrid og hydrogenperoksid.
Spesielt populært firma m. -Hlorperbenzoesyre, siden det er relativt trygt i omløp, er det ganske stabilt og kan lagres i lang tid.
Oksidasjon oppstår på grunn av det frigjorte oksygenatomet:
Rcoooh \u003d rcoh + [o]
Nadcisitles brukes til å oppnå epoksyder fra alkener, samt laktoner fra alicykliske ketoner.
Hydrogenperoksid
Hydrogenperoksid er en fargeløs væske, blandet med vann, etanol og dietyleter. En 30% løsning H 2 O 2 kalles perhydro. Det høyt konsentrerte stoffet kan reagere med organiske stoffer med en eksplosjon. Når du lagrer dekomponer på oksygen og vann. Motstanden for hydrogenperoksid øker med fortynning. For oksidasjon anvendes vandige løsninger av forskjellige konsentrasjoner (fra 3 til 90%) i nøytrale, sure eller alkaliske medier.
H20 2 \u003d H 2 O + [O]
Handlingen av dette reagenset på α, β-ubebodde karbonylforbindelser i det alkaliske medium oppnås ved passende epoksyaldehyder og ketoner, oksydasjonen av karboksylsyrer i det sure medium syntetiseres. En 30% løsning H202 i eddiksyre oksiderer alkener i 1,2-diol. Hydrogenperoksid brukes: for å oppnå organiske og uorganiske peroksyder, pebble og perkarbonat Na; som et oksiderende middel i rakettbrensel; i fremstillingen av epoksyder, hydrokinon, pyrocatechin, etylenglykol, glyserin, vulkaniseringsakseleratorer av gruppen av Tyurama et al.; for bleking av oljer, fett, pels, lær, tekstilmaterialer, papir; for rengjøring germanium og silisium halvleder materialer; som desinfeksjonsmiddel for avhending av husholdning og industriell avløpsvann; i medisin; som en kilde på 2 i ubåter; H202 er en del av Fenton-reagenset (Fe 2 + H202), som brukes som en kilde til frie radikaler i organisk syntese.
Ruthenium tetraxides og osmia
Tetraoksid Osmia OSO 4 - Pulver fra hvitt til blekgul farge med t. Pl. 40,6ºС; t. kip. 131.2ºс Den fjernes ved romtemperatur, løselig i vann (7,47 g i 100 ml ved 25 ° C), CCl4 (250 g 100 g løsningsmiddel ved 20 ° C). I nærvær av organiske forbindelser, på grunn av gjenopprettelsen til OSO 2.
Ruo 4 er en gylden gul prisme med t. Pl. 25.4ºс, det er merkbart oppnådd ved romtemperatur. Moderat oppløselig i vann (2,03 g per 100 ml ved 20 ° C), meget godt løselig i CCl 4. Sterk oksidasjonsmiddel enn OSO 4. Over 100ºс eksploderer. I tillegg til osmium tetraoksid har stor toksisitet og høy pris.
Disse oksidasjonene brukes til oksydasjon av alkener i a-glykoler i milde forhold.
Dette materialet kan foldes i uttrykk i selvstudium, på grunn av den store mengden informasjon, mange nyanser, alle slags, men hvis. Les nøye!
Hva vil vi bare snakke om?
I tillegg til fullstendig oksidasjon (forbrenning), for enkelte klasser av organiske forbindelser, karakteriseres reaksjonen av ufullstendig oksidasjon, mens de blir til andre klasser.
Det er spesifikke oksidasjonsmidler for hver klasser: CUO (for alkoholer), CU (OH) 2 og OH (for aldehyder) og andre.
Men det er to klassiske oksidasjonsmidler som, hvis så uttrykkes, universell for mange klasser.
Dette er kaliumpermanganat - KMNO 4. Og en bichromat (dikromat) av kalium - K 2 Cr 2 o 7. Disse stoffene er sterke oksidasjonsmidler på grunn av mangan i graden av oksidasjon +7 og krom til graden av oksidasjon +6, henholdsvis.
Reaksjoner med disse oksidasjonene er funnet ganske ofte, men ingen steder er det ikke noe holistisk lederskap, ifølge hvilket prinsipp velger produkter av slike reaksjoner.
I praksis er det mange faktorer som påvirker reaksjonen (temperatur, middels, konsentrasjon av reagenser, etc.). Ofte viser det seg en blanding av produkter. Derfor, for å forutsi produktet som dannes nesten umulig.
Og for eksamen, er det ikke egnet: det er umulig å skrive "kanskje eller så, eller så eller på annen måte eller en blanding av produkter." Det trenger en spesifikk.
Oppgavekompilatorene har investert en bestemt logikk, et bestemt prinsipp som skal skrive et bestemt produkt. Dessverre delte de ikke med noen.
Dette spørsmålet i de fleste fordeler er ganske glidende av festen: to eller tre reaksjoner er gitt som et eksempel.
Jeg presenterer i denne artikkelen, hva kan kalles resultatene av forskningsanalysen av bruksoppgavene. Logikken og prinsippene for kompilering av oksidasjonsreaksjoner permanganat og solidaritets dikromat er ganske høy nøyaktighet (i samsvar med f.eks. Standarder). Om alt i orden.
Bestemmelse av oksidasjon.
Den første når vi har å gjøre med redoksreaksjoner, er det alltid et oksidasjonsmiddel og reduksjonsmiddel.
Oxidisator er en mangan i permanganat eller krom i et dikromat, reduksjonsmiddel - atomer i et organisk (nemlig karbonatomer).
Lite å identifisere produkter, må reaksjonen utlignes. For justeringen, bruk tradisjonelt elektronisk balanse metode. For å anvende denne metoden, er det nødvendig å bestemme graden av oksidasjon av reduksjonsmidler og oksidasjonsmidler før og etter reaksjonen.
Uorganiske stoffer i graden av oksidasjon kan være i stand til å grave 9:
Men i organisasjonen ble det sannsynligvis ikke bestemt i klasse 9. Derfor, før du lærer å skrive OSR i organisk kjemi, må du lære å bestemme graden av karbonoksydasjon i organiske stoffer. Det er gjort litt annerledes, ellers enn i uorganisk kjemi.
Karbon har maksimal grad av oksidasjon +4, minimum -4. Og det kan vise noen grad av oksidasjon av dette gapet: -4, -3, -2, -1, 0, + 1, +2, +3, +4.
Først må du huske hva som er graden av oksidasjon.
Graden av oksidasjon er en betinget ladning som oppstår på atomet, når det antas at elektroniske par blir skiftet helt mot et mer elektronegativt atom.
Derfor er graden av oksidasjon bestemt av antall fordrevne elektroniske par: Hvis det forskyves til dette atomet, kjøper den en overdreven minus (-) ladning, hvis fra et atom, så kjøper det en overdreven pluss (+) ladning. I prinsippet er dette hele teorien du trenger å vite for å bestemme graden av oksidasjon av karbonatomet.
For å bestemme graden av oksidasjon av et bestemt karbonatom i forbindelsen, må vi vurdere hver forbindelse og se hvilken retning elektrondampen vil bli skiftet og hvilken overskuddsladning (+ eller -) vil skyldes dette på karbonatomet.
Vi vil analysere spesifikke eksempler:
Karbon tre bånd med hydrogen. Karbon og hydrogen - hvem er mer elektronerbar? Kull, det betyr, på disse tre obligasjonene, vil det elektroniske paret skifte mot karbon. Karbon tar hver hydrogen for en negativ ladning: det viser seg - 3
Fjerde forbindelse med klor. Karbon og klor - hvem er mer elektronerbar? Klor, det betyr, i denne forbindelse, vil det elektroniske paret skifte mot klor. Karbonet vises en positiv ladning +1.
Deretter trenger du bare å brette: -3 + 1 \u003d -2. Graden av oksidasjon av dette karbonatomet: -2.

Vi definerer graden av oksidasjon av hvert karbonatom:

Kull har tre bånd med hydrogen. Karbon og hydrogen - hvem er mer elektronerbar? Kull, det betyr, på disse tre obligasjonene, vil det elektroniske paret skifte mot karbon. Karbon tar hver hydrogen for en negativ ladning: det viser seg - 3
Og en annen forbindelse med et annet karbon. Kull og annet karbon - deres elektronegabilitet er like, slik at den elektroniske parforskyvningen ikke forekommer (forbindelsen er ikke polar). 

Dette atomet har to bånd med ett oksygenatom, og et annet forhold til et annet oksygenatom (som en del av OH-gruppen). Flere elektronegative oksygenatomer i tre bindinger forsinker det elektroniske paret fra karbon, karbonet vises +3.
Den fjerde tie karbon er forbundet med et annet karbon, som vi allerede har snakket, på denne forbindelse er elektronparet ikke skiftet.


To obligasjoner, karbon er forbundet med hydrogenatomer. Karbon, da jo mer elektonegativ forsinket seg på ett par elektroner for hver forbindelse med hydrogen, kjøper en ladning -2.
Dobbel avhengighet av karbon er forbundet med et oksygenatom. Flere elektronegative oksygendenserer ett elektronisk par for hver forbindelse. Sammen viser seg i karbon, to elektroniske par er forsinket. Carbon kjøper +2 kostnad.
Sammen viser det seg +2 -2 \u003d 0.

Vi definerer graden av oksidasjon av dette karbonatomet:
Trippelforholdet med mer elektronegativ nitrogen - gir karbonavgift +3, på grunn av karbon-bias-karbonet forekommer ikke.

Oksidasjon permanganat.
Hva skjer med permanaganat?
Redoksreaksjon med permanganat kan strømme i forskjellige medier (nøytral, alkalisk, syre). Og det avhenger av miljøet, da det er at reaksjonen vil strømme, og hvilke produkter dannes.
Derfor kan det gå i tre retninger:

Permanganat, som er en oksidasjonsmiddel, gjenopprettes. Her er produktene i gjenopprettingen:
- Aclest medium.
Onsdag surgjøres med svovelsyre (H2SO4). Manganet gjenopprettes til graden av oksidasjon +2. Og gjenopprettingsprodukter vil være:
KMNO 4 + H 2 SO 4 → MNSO 4 + K2SO4 + H 2 O
- Alkalisk miljø.
For å skape et alkalisk medium tilsettes en ganske konsentrert alkali (KOH). Manganet gjenopprettes til graden av oksidasjon +6. Gjenopprettingsprodukter
KMNO 4 + KOH → K 2 MNO 4 + H 2 O
- Nøytralt miljø (og svakt alkalo).
I det nøytrale mediumet, i tillegg til permanganat, kommer vann også i reaksjonen (som vi skriver på venstre side av ligningen), mangan vil bli gjenvunnet til +4 (MNO 2), gjenopprettingsproduktene vil være:
KMNO 4 + H 2 O → MNO 2 + KOH
Og i et litt alkalisk medium (i nærvær av en løsning av KOH lav konsentrasjon):
KMNO 4 + KOH → MNO 2 + H 2 O

Hva skjer med organisk?
Det første du trenger å lære er alt som starter med alkohol! Dette er det opprinnelige stadiet av oksidasjon. Oksydasjonen underkastes det karbonet som hydroksylgruppen er festet til.
Under oksidasjonen kjøper karbonatomet "et bånd med oksygen. Derfor, når oksidasjonsreaksjonsskjemaet er skrevet, er [O] skrevet over pilen:
Primær alkohol Oksider først til aldehyd, deretter til karboksylsyre:

Oksidasjon sekundær alkohol Ut i andre trinn. Siden karbon er en overgivelse, dannes keton, og ikke aldehyd (karbonatom i ketongruppen er allerede fysisk ikke i stand til å danne et bånd med en hydroksylgruppe):

Ketoner, tertiære alkoholer og karboksylsyrer Neste oksideres ikke lenger:

Prosessen med oksidasjonstrinnet - mens det er der det skulle oksidere, og det er alle forhold for dette - reaksjonen kommer. Alt slutter med produktet som i disse forholdene ikke oksideres: tertiær alkohol, keton eller syre.
Det er verdt å merke seg scenen av oksidasjon av metanol. I tillegg oksyderes det til det passende aldehydet, deretter til den aktuelle syre:

Specularity av dette produktet (myresyre) er at karbon i karboksylgruppen er forbundet med hydrogen, og hvis du ser nærmere, kan du se at det ikke er noe mer enn aldehydgruppe:

Og aldehydgruppen, som vi fant ut tidligere, oksyderes til karboksyl:

Anerkjente det resulterende stoffet? Dens brutto formel h 2 co 3. Dette er kullsyre som desintegrerer karbondioksid og vann:
H 2 CO 3 → H 2 O + CO 2
Derfor oksideres metanol, myr aldehyd og myresyre (på bekostning av aldehydgruppen) til karbondioksid.
Myk oksidasjon.
Myk oksidasjon er oksidasjon uten sterk oppvarming i nøytral eller litt alkalisk medium (over reaksjonen er skrevet 0 ° eller 20. °) .
Det er viktig å huske at alkoholer ikke oksideres i milde forhold. Derfor, hvis de er dannet, så oksydasjon og stopper. Hvilke stoffer vil bli med i reaksjonen av myk oksidasjon?
- Inneholdende dobbeltbinding C \u003d C (Wagner-reaksjon).
Samtidig er π-bindingen brutt og de frigjorte bindingene "sitter" på hydroksylgruppen. Det viser seg en dobbel alkohol:

Vi skriver reaksjonen av den myke oksydasjonen av etylen (eten). Vi skriver utgangsmaterialene og forutsier produktene. Samtidig skriver H 2 O og COH ikke ennå: De kan vise seg å være både i den rette delen av ligningen og til venstre. Og umiddelbart bestemme graden av oksidasjon av deltakende stoffer:

Vi vil lage en elektronisk balanse (vi mener at reduksjonsmidlet er to - to karbonatomer, de oksyderes separat):

Velg koeffisientene:

Til slutt er det nødvendig å legge til manglende produkter (H20 og KOH). Den rette mangler kalium - det betyr at alkalien vil være riktig. Vi legger koeffisienten foran den. Til venstre er det ikke nok hydrogen, det betyr at vannet er igjen. Vi legger koeffisienten foran den:

Vi gjør det samme med propylen (propan):


Passer ofte til cykloalkan. La ham ikke skremme deg. Dette er et konvensjonelt hydrokarbon med dobbeltbinding:

Hvor denne doble forbindelsen var, vil oksydasjonen gå likt:

- Inneholdende aldehydgruppe.
Aldehydgruppen er mer reaktiv (det er lettere å reagere) enn alkohol. Derfor vil aldehydet oksyderes. Å syre:

Tenk på eksemplet på acetaldehyd (etanle). Vi skriver reagenser og produkter og legger graden av oksidasjon. Vi vil balansere og sette koeffisientene før reduksjonsmiddelet og oksiderende middel:

I det nøytrale mediumet og det litt alkaliske kurset av reaksjonen vil være litt annerledes.
I det nøytrale mediumet, som vi husker, på venstre side av ligningen, skriver vi vann, og i den rette delen av alkali-ligningen (dannet under reaksjonen):

I dette tilfellet finnes syre og alkali i en blanding. Nøytralisering oppstår.

De kan ikke eksistere nær og reagere, saltformer:
På samme tid, hvis vi ser på koeffisientene i ligningen, vil vi forstå at syrene er 3 bønn, og alkali 2 bønn. 2 Bønn alkali kan nøytralisere bare 2 bønnesyrer (2 bønnsalter dannes). Og en mølle av syre forblir. Derfor vil den endelige ligningen være slik:

I det litt alkaliske medium er det alkali i overskudd - det tilsettes til reaksjonen, så all syre er nøytralisert:

En lignende situasjon oppstår når metanalen oksyderes. Han, som vi husker, oksyderes til karbondioksid:

Det bør tas i betraktning at karbonoksyd (IV) CO 2 er sur. Og vil reagere med alkali. Og siden kullsyren er en to-akse, kan dannes både surt salt og gjennomsnitt. Det avhenger av forholdet mellom alkali og karbondioksid:
Hvis alkalien refererer til karbondioksid som 2: 1, så vil det være et gjennomsnittlig salt:
Eller alkali kan være betydelig mer (mer enn to ganger). Hvis det er mer enn to ganger, vil balansen mellom alkalier forbli:
3koh + CO 2 → K 2 CO 3 + H 2 O + KOH
Dette vil forekomme i et alkalisk medium (hvor alkalioverskudd, som den tilsettes til reaksjonsblandingen til reaksjonen) eller i et nøytralt miljø når alkalien dannes mye.
Men hvis alkalien refererer til karbondioksid som 1: 1, det blir et surtalt:
KOH + CO 2 → KHCO 3
Hvis karbondioksid er større enn du trenger, forblir den overskytende:
KOH + 2CO 2 → KHCO 3 + CO 2
Dette vil være i et nøytralt miljø hvis alkali dannes ikke nok.
Vi skriver kildestoffene, produktene, vi vil balansere balansen mellom oksidasjon foran oksidasjonsmiddelet, reduksjonsmiddel og produkter som dannes av dem:

I det nøytrale medium til høyre vil danne alkali (4koh):

Nå er det nødvendig å forstå hva som skal dannes av samspillet mellom tre mol CO 2 og fire alkali mol.
3CO 2 + 4KOH → 3KHCO 3 + KOH
KHCO 3 + KOH → K 2 CO 3 + H 2 O
Derfor viser det seg slik:
3CO 2 + 4KOH → 2KHCO 3 + K 2 CO 3 + H 2 O
Derfor, i den rette delen av ligningen, skriver vi to bønnepor og en mol karbonat:

Og i et svakt alkalisk miljø er det ingen slike problemer: på grunn av at alkalioverskudd vil bli dannet av gjennomsnittlig salt:

Det samme vil være ved oksydasjonen av aldehydet av oksalsyre:

Som i det foregående eksempel dannes en dibasinsyre, og 4 bønn alkali bør være på ligningen (som 4 bønn permanganat).
I et nøytralt miljø igjen er hele alkalien ikke nok for den fullstendige nøytraliseringen av hele syren.
Tre bønnalkalier går på dannelsen av surt salt, en mol alkali forblir:
3HOOC-COOH + 4KOH → 3KOOC-COOH + KOH
Og denne mol alkali går i samspill med et enkelt syre salt:
KOOC-COOH + KOH → KOOC-COOK + H 2 O
Det viser seg slik:
3HOOC-COOH + 4KOH → 2KOOC-COOH + KOOC-COOK + H 2 O
Sluttligning:

I det svakt alkaliske medium dannes gjennomsnittlig salt på grunn av overskytende alkali:

- Inneholdende trippelforholdetC.≡ C..
Husk hva som var med en myk oksidasjon av forbindelser med en dobbeltbinding? Hvis du ikke husker, så bla tilbake - husk.
Den π-bindingen er sprut, karbonatomene er festet langs hydroksylgruppen. Her er det samme prinsippet. Det er bare verdt å huske at det er to π-obligasjoner i trippelbinding. Først skjer det på den første π-kommunikasjonen:
Deretter på en annen π-kommunikasjon:

Strukturen der ett karbonatom har to hydroksylgrupper, ekstremt ustabile. Når noe ikke er opprettholdt i kjemi, søker den å "falle av". Vann faller av, slik:

Karbonylgruppen er oppnådd.
Vurder eksempler:
Etin (acetylen). Tenk på stadiene av oksidasjon av dette stoffet:

Vannrenser:




Som i det foregående eksempel, i en enkelt reaksjonsblanding av syre og alkali. Nøytralisering oppstår - saltformer. Som det kan ses av koeffisienten før permanganat, vil alkalien være 8 mol, det vil si det er nok for nøytralisering av syre. Sluttligning:

Vurder Boudin-2 oksidasjon:

Vannrenser:

Her er syre ikke dannet, så det er ikke nødvendig å lure over nøytralisering.
Reaksjonsligning:

Disse forskjellene (mellom karbonoksydasjon med kanten og i midten av kjeden) er sterkt demonstrert av eksempelet på pentina:

Vannrenser:

Det viser seg et stoff av en interessant struktur:

Aldehydgruppen fortsetter å oksidere:
Vi skriver utgangsmaterialene, produktene som bestemmer grader av oksidasjon, vi vil bli balansert, enkle koeffisienter før oksidasjonsmiddel og reduksjonsmiddel:

Alkali skal danne 2 hauger (siden koeffisienten før permanganat 2), derfor er hele syren nøytralisert:

Hard oksidasjon.
Hard oksidasjon er oksidasjon i sur, eliminere Medium. Så vel som i nøytral (eller lav-alkalisk), men når den er oppvarmet.
I det sure mediumet, oppvarmet også. Men at hard oksidasjon går ikke i et surt medium, er oppvarming en forutsetning.
Hvilke stoffer vil bli utsatt for hard oksidasjon? (I utgangspunktet vil vi bare analysere i et surt miljø - og deretter legge til nyanser som oppstår når de oksiderende i en sterkt alkoholisk og nøytral eller svakt alkalisk (når oppvarmet) medium).
Med hard oksidasjon, går prosessen til maksimum. Mens det er at oksidert oksidasjon går.
- Alkoholer. Aldehyder.
Tenk på oksydasjonen av etanol. Den oksyderes til syren:

Registreringsligning. Vi skriver ned startmaterialene, produktene i OSR, setter graden av oksidasjon, utgjør balansen. Juster reaksjonen:

Hvis reaksjonen utføres ved kokepunktet til aldehyd når den er å danne, vil den fordampe (flyve bort) fra reaksjonsblandingen, ikke så mye å oksidere. Den samme effekten kan oppnås i svært milde forhold (svak oppvarming). I dette tilfellet skriver vi aldehyd som et produkt:

Tenk på oksydasjonen av den sekundære alkoholen på eksempelet på propanol-2. Som allerede nevnt, er oksydasjon brutt i andre trinn (dannelsen av karbonylforbindelse). Siden keton dannes, som ikke oksideres. Reaksjonsligning:

Oksidasjon av aldehyder anser ved temaet etanale. Det oksyderes også til syren:

Reaksjonsligning:

Metanal og metanol, som nevnt tidligere, oksydert til karbondioksid:

Metanal:

- Inneholdende flere obligasjoner.
I dette tilfellet bryter kjeden over flere kommunikasjoner. Og atomer som har dannet det oksyderes (skaffe en binding med oksygen). Oksider så langt som mulig.
Når dobbeltbindingen bryter fra utklippene, dannes karbonylforbindelser (i diagrammet nedenfor: fra en burst - aldehyd, fra den andre - Keton)

![]()
Vi vil analysere oksydasjonen av Penten-2:

Oksidasjon av "scraps":

Det viser seg at to syrer dannes. Vi skriver utgangsmaterialene og produktene. Vi definerer grader av oksidasjon i atomer, som endrer den, balansert, utjevner reaksjonen: 
Ved å utgjøre den elektroniske balansen mener vi at reduksjonsmidlet er to-to karbonatomer, de oksyderes separat:

Syre vil ikke alltid danne. Vi vil for eksempel analysere oksidasjon av 2-metylbuteten:

Reaksjonsligning:

Helt samme prinsipp når oksiderende forbindelser med en trippelbinding (bare oksydasjon er umiddelbart med dannelsen av en syre, uten en mellomliggende formasjon av aldehyd):

Reaksjonsligning:

Når en flere tilkoblinger er plassert nøyaktig i midten, så ikke to produkter, men en. Siden "scraps" er de samme, og de oksyderes til de samme produktene:

Reaksjonsligning:

- To ganger krone syre.
Det er en syre som karboksylgrupper (kroner) er koblet til hverandre:
Dette er sorvalsyre. De to kronene er vanskelige å komme sammen. Det er absolutt stabilt under normale forhold. Men på grunn av det faktum at i det er to karboksylgrupper forbundet med hverandre, er det mindre stabilt enn andre karboksylsyrer.
Og derfor, med spesielt tøffe miljøer, kan det oksyderes. Det er en pause mellom de "to kronene":

Reaksjonsligning:

- Gomologer av benzen (og deres derivater).
Benzol selv oksyderes ikke, på grunn av det faktum at aromatikkene gjør denne strukturen veldig stabil 
Men hans homologer oksyderes. Samtidig bryter kjeden, det viktigste er å vite hvor nøyaktig. Det er noen prinsipper:
- Benzenringen i seg selv ødelegger ikke, og forblir så langt til slutten, oppstår brudd på kommunikasjon i det radikale.
- Oksiserer et atom direkte relatert til benzenringen. Hvis, etter det, fortsetter karbonkjeden i radikalet - så vil gapet være etter det.


Vi vil analysere oksydasjonen av metylbenzen. Det er ett karbonatom i radikalet oksiderer: 
Reaksjonsligning:

Vi vil analysere oksydasjonen av isobutylbenzenet:

Reaksjonsligning:

Vi vil analysere oksydasjonen av det andre butylbenzenet:

Reaksjonsligning:

I oksydasjonen av gomologer av benzen (og derivater av homologer) med flere radikaler, dannes to-tre og flere store aromatiske syrer. For eksempel, oksidasjon av 1,2-dimetylbenzen:

Derivatene til homologene av benzen (hvor benzenringen ikke er hydrokarbonradikaler), oksideres det samme. En annen funksjonell gruppe forstyrrer ikke benzenringen:


Subtotal. Algoritme "Slik registrerer du svaret på hard oksidasjon permanganat i et surt miljø":
- Registrer startmaterialer (organisk + kmno 4 + H 2 så 4).
- Skriv organiske oksidasjonsprodukter (forbindelser som inneholder alkohol, aldehydgrupper, flere obligasjoner, samt benzenhomologer).
- Skriv per(MNSO 4 + K2SO4 + H20).
- Bestemme grader av oksidasjon fra deltakerne i OSR. Balansere. Slip koeffisientene til oksidasjonsmiddelet og reduksjonsmidlet, så vel som stoffene som dannes av dem.
- Deretter anbefales det å beregne hvor mange sulfatanioner i den rette delen av ligningen, i samsvar med dette setter koeffisienten foran svovelsyren til venstre.
- På slutten, legg koeffisienten før vann.
Hard oksidasjon i en sterkt alternativ medium og nøytral eller svakt alkalisk (når oppvarmet).
Disse reaksjonene er mye mindre vanlige. Det kan sies at slike reaksjoner er eksotiske. Og som det skal gjøres av noen eksotiske reaksjoner, var disse de mest motstridende.
Hard oksidasjon Det er stivt i Afrika, så det organiske oksyderes på samme måte som i et surt miljø.
Separat vil vi ikke demontere reaksjonen for hver klasse, siden det generelle prinsippet allerede er skissert tidligere. Vi vil bare analysere nyansene.
Eliminere miljøet :
I en sterkt alger gjenopprettes permanganat til graden av oksidasjon +6 (kaliumsmangan):
KMNO 4 + KOH → K 2 MNO 4.
I en sterk alkalialkali er det alltid et overskudd, så fullstendig nøytralisering vil bli avholdt: hvis karbondioksid dannes - det vil være et karbonat hvis syre dannes - det vil være et salt (hvis syren er midtsaltet).
For eksempel, oksydasjonen av propen:

Etylbenzenoksydasjon:

Etikett eller nøytralt medium når det er oppvarmet :
Det må også alltid ta hensyn til muligheten for nøytralisering.
Hvis oksidasjon strømmer i et nøytralt medium og en sur forbindelse (syre eller karbondioksid) dannes, vil den resulterende alkohol nøytralisere denne syreforbindelsen. Men ikke alltid alkali er nok for fullstendig nøytralisering av syre.
Når aldehydene oksyderes, er det for eksempel ikke nok (oksidasjon vil fortsette på samme måte som i milde forhold - temperaturen akselererer bare reaksjonen). Derfor dannes salt og syre (de resterende grovt tale i overskudd).
Vi diskuterte det når de demonterer den myke oksydasjonen av aldehyder.
Derfor, hvis syren er dannet i det nøytrale miljøet, må du se nøye ut hvis det er nok for nøytralisering av hele syren. Spesiell oppmerksomhet bør betales til nøytraliseringen av polypiske syrer.
I et litt alkalisk medium dannes bare mediumsalter på grunn av tilstrekkelig antall alkali, som alkalioverskudd.
Som regel er alkali under oksidasjon i et nøytralt medium nok. Og reaksjonsligningen som er nøytral at i det svakt alkaliske mediet vil være det samme.
For eksempel vil vi analysere oksydasjonen av etylbenzen:


Alkali er nok for den komplette nøytraliseringen av de sure forbindelsene, til og med overskudd forblir:

3 Bønn Alkali forbrukes - 1 gjenstander.
Sluttligning:

Denne reaksjonen i et nøytralt og lavt alkalisk medium vil gå likt (det er ingen alkali i lavt alkalisk medium, men dette betyr ikke at det ikke er, det kommer ikke inn i reaksjonen).
Redox reaksjoner som involverer dikromatet (bichromat) av kalium.
Bichromat har ikke så stort utvalg av organiske oksidasjonsreaksjoner i eksamen.
Oksidasjon av Bichromat utføres vanligvis bare i et surt miljø. Med dette krom blir gjenopprettet til +3. Gjenopprettingsprodukter:
Oksidasjon vil være tøft. Reaksjonen vil være svært lik oksydasjonen av permanganat. De samme stoffene vil oksidere at de oksyderes av permanganat i et surt miljø, vil de samme produktene bli dannet.
Vi vil analysere noen reaksjoner.
Tenk på oksydasjonen av alkohol. Hvis vi utfører oksydasjon ved kokepunktet til aldehyd, vil den kjære sin reaksjonsblanding uten utsatt for oksidasjon:

Ellers kan alkoholen oksyderes direkte til syren.
Aldehyd, oppnådd i den forrige reaksjonen, kan "fanges", og gjøre det oksydert til syre:

Oksidasjon av cykloheksanol. Cyclohexanol er en sekundær alkohol, så keton dannes:

Hvis det er vanskelig å bestemme graden av oksidasjon av karbonatomer med en slik formel, kan males på utkastet:

Reaksjonsligning:

Vurder oksydasjonen av Cyclopenten.
Dobbelbindingen bryter (syklusen åpnes), atomene som danner det, oksyderes til maksimum (i dette tilfellet til karboksylgruppen):


Noen funksjoner i oksidasjon i eksamen, med hvem vi ikke helt er enige om.
De "reglene", prinsipper og reaksjoner som vil bli diskutert i denne delen, vurderer vi ikke helt riktig. De motsier seg ikke bare den virkelige situasjonen (kjemi som vitenskap), men også intern logikk av skoleprogrammet og eksamen spesielt.
Men likevel er vi tvunget til å gi dette materialet nøyaktig i skjemaet som eksamenen krever.
Det vil være om hard oksidasjon.
Husk hvordan gomologer oksyderes og deres uttalt i tøffe forhold? Radikaler er helt ødelagte - karboksylgrupper dannes. Skrapene er utsatt for oksidasjon allerede "uavhengig":

Så, hvis en hydroksylgruppe plutselig er et radikalt, eller en flere kommunikasjon, må du glemme at det er en benzenring. Reaksjonen vil bare gå på denne funksjonelle gruppen (eller flere kommunikasjoner).
Funksjonell gruppe og flere kommunikasjonsgaver enn benzenring.

Vi vil analysere oksydasjonen av hvert stoff:
Første stoff:

Du må ikke være oppmerksom på at det er en benzenring. Fra bruken av bruken er bare sekundær alkohol. Sekundære alkoholer oksyderes til ketoner, og ketoner oksyderes ikke:

La dette stoffet oksyderes av bichromat:

Andre stoffet:

Dette stoffet oksyderes, bare som en dobbeltsidig forbindelse (vi tar ikke hensyn til benzenringen):

La det oksyderes i nøytral permanganat når det er oppvarmet:

Dannet alkali nok til fullstendig nøytralisering av karbondioksid:
2koh + CO 2 → K 2 CO 3 + H 2 O
Total ligning:

Oksidasjon av det tredje stoffet:

Anta at oksidasjon vil lekke kalium permanganat i et surt miljø:

Oksidasjon av fjerde sak:

Den oksyderes i strongshop-mediet. Reaksjonsligningen vil være:

Vel, til slutt, det er så oksydert av vinylbenzen:

Og det oksyderes til benzoesyre, det er nødvendig å huske på at i henhold til eksamens logikk er det så oksidert fordi det er et derivat av benzen. Og fordi den inneholder en dobbeltbinding.
Konklusjon.
Det er alt du trenger å vite om redoksreaksjoner som involverer permanganat og bichromat i en organisk.
Ikke bli overrasket om, noen øyeblikk som er angitt i denne artikkelen, hører du for første gang. Som allerede nevnt, er dette emnet svært omfattende og motstridende. Og til tross for dette, av en eller annen grunn, betaler hun ekstremt liten oppmerksomhet.
Som du kanskje har vært overbevist, forklarer to-tre reaksjoner ikke alle mønstrene av disse reaksjonene. Her trenger du en integrert tilnærming og en detaljert forklaring på alle øyeblikk. Dessverre i lærebøkene og på Internett-ressursene, er emnet ikke fullt ut avslørt, eller ikke avslørt i det hele tatt.
Jeg prøvde å eliminere disse feilene og manglene og vurdere dette emnet helt, og ikke delvis. Jeg håper jeg klarte det.
Takk for din oppmerksomhet, dere alle er gode! Suksesser i utviklingen av kjemisk vitenskap og bestått eksamener!
4.5. Oksidasjon av alkener
Alkenxideringsreaksjoner er tilrådelig å dele seg i to store grupper: Reaksjoner hvor karbonskjelettet og reaksjonen av den oksidative ødeleggelse av karbonskjelettet av dobbeltbindingsmolekylet er bevart. Den første gruppe reaksjoner inkluderer epoksydasjon, så vel som hydroksylering, noe som resulterer i dannelsen av vicinale dioler (glykoler). I tilfelle av sykliske alkener under hydroksylering dannes vicinal transe- eller cIS.-Dioles. En annen gruppe inkluderer ozonolyse og en reaksjon av en uttømmende oksidasjon av alkener, som fører til dannelsen av forskjellige typer karbonylforbindelser og karboksylsyrer.
4.5.a. Oksydasjonsreaksjonene av alkener samtidig som det opprettholdes et karbonskjelett
1. Epoxidasjon (reaksjon N.A. Prilezhev, 1909 g)
Aciclic og cykliske alkener når de samhandler med peracid (Suprachlotes) RCOOOOH i en ikke-polar, likegyldig mediumformet epoksyder (oksiranere), slik at reaksjonen i seg selv kalles epoksidasjonsreaksjonen.
Ifølge moderne nomenklatur Jupak. - En tremedlem syklus med ett oksygenatom kalles Oxira.
Epoksydasjon av alkener bør betraktes som en synkron, koordinert prosess der ioniske mellomprodukter av typen hydroksylkation ikke er involvert. Med andre ord er epoxidasjonen av alkenes en prosess syn- Troubleto ett oksygenatom for dobbeltkommunikasjon med full bevaring av substituentkonfigurasjonen ved dobbeltbinding.
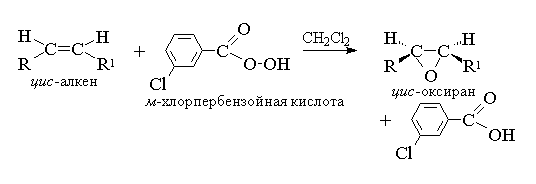

For epoksydasjon ble det foreslått en mekanisme for avtalt prosesser.

T. K. Dobbeltbindingsangrepatom av oksygenoksygen er like lik begge sider av dobbeltbindingsplanet, dannede oksyraner er heller meso.- skjemaer, eller blandinger av enantiomerer. Følgende peculoter brukes som epoxideringsmidler: Pebenzoic, m.-Hlorperbenzoic, Monoperfthal, Perussum, Trifluorperuce og Permanteury. Perkislotene av de aromatiske serien brukes i form av individuelle reagenser, mens perkislotes av alifatiske rader - CH3C03H, CF333 og NSO3 n er ikke isolert individuelt, men brukes etter deres formasjon under samspillet på 30% eller 90% hydrogenperoksid og passende karboksylsyre. Perbenzoic I. m.-Hlorperbenzosyre oppnås ved henholdsvis oksidasjon, benzoisk og m.-Hlorbenzosyrer med 70% hydrogenperoksid i metansulfonsyreoppløsning eller fra kloridhydrhydrider av disse syrene og hydrogenperoksidet.


Monoperftalsyre oppnås ved en lignende fremgangsmåte fra ftalalanhydrid og 30% hydrogenperoksid.

I utgangspunktet ble Pebenzoic eller Monoperftinsyrer brukt til å oppnå oksiraner (epoksider):

For tiden, for epoksydasjon, bruker oftest m.-Hlorperbenzosyre. I motsetning til andre perkslot er det stabilt når det lagres i lang tid (opptil 1 år) og er helt sikker når man kontakter. OxyRants oppnådd under oksydasjonen av acykliske og cykliske alkener m.-Herperbenzosyre i metylenkloridoppløsning, kloroform eller dioksan, vanligvis ganske høy.



Perkislotes genereres ofte direkte i reaksjonsblandingen på 90% hydrogenperoksid og karboksylsyre i metylenklorid.


Double-bind alkenes assosiert med en karbonylgruppe eller en annen akseptor substituent, lavaktiv og for oksidasjon er det bedre å bruke sterkere oksidanter, så som trifluoroprucus syre oppnådd fra trifluoreddiksyreanhydrid og 90% hydrogenperoksid i metylenklorid. Det enkleste oksiran-etylenoksydet er oppnådd i industrien ved oksydasjon av etylen ved oksygen i nærvær av sølv, som en katalysator.

2. anti.-Hhydroksylering
Den tre-ledede ringen av oksyraner er lett avslørt under virkningen av et bredt utvalg av nukleofile reagenser. Disse reaksjonene vil bli diskutert i detalj i seksjonen dedikert til acyklisk og syklisk enkel eter. Bare Oxyranes hydrolyse vil bli vurdert her. Hydrolysen av oksyraner katalyseres av både syrer og baser. I begge tilfeller dannes vicinal dioler, dvs. glykoler. I syre katalyse i første fase er oksygenoksydatomet protonert til dannelsen av en cyklisk oksoniumkation, som er beskrevet som et resultat av et nukleofilt angrep av vannmolekylet:

Nøkkelfasen i beskrivelsen av ringen, som bestemmer hastigheten på hele prosessen er det nukleofile angrepet med vann på den protonerte dannelsen av oksiran. Fra mekanismenes synspunkt ligner denne prosessen på beskrivelsen av en bromoniumion med et nukleofilt angrep av et bromidion eller et annet nukleofilt middel. Fra disse stillingene bør stereokjemiske resultater være utdanning transe- Glykoler ved splitting av sykliske epoksyder. Faktisk, med den syre-katalyserte hydrolysen av cykloheksenoksid eller cyklopenthenoksyd, dannes eksklusivt transe-1,2-diol.

Således tilsvarer den to-trinns prosessen med epoksydasjon av alken etterfulgt av syrehydrolyse av epoksid totalt til reaksjonen anti.- Hydroksilisering av alkener.
Begge stadier anti.- Hydroksylinering av alkener kan kombineres dersom alken blir behandlet med vandig 30-70% hydrogenperoksid i maur eller trifluoreddiksyre. Begge disse syrene er sterke nok til å forårsake opplysning av oksiransyklusen.

Opplysningen av oksyringsringen som er katalysert av basen, fører også til dannelsen av cyklisk transe-Glikoli.

Følgelig er den to-trinns prosessen med epoksydasjon av alkener med etterfølgende alkalisk hydrolyse av epoksyder også en reaksjon anti.- Hydroksilisering av alkener.
3. syn-Hhydroksylering
Noen salter og oksider av overgangsmetaller i de høyeste grader av oksidasjon er effektive reagenser. syn- Hydroksilisering av dobbeltbindingen til alkenet når begge hydroksylgruppene er forbundet med samme side av den dobbelte forbindelsen. Oksidasjon av alkener permanganatkalium - en av de eldste metodene syn- Hydroksilisering av dobbeltkommunikasjon - fortsetter å bli brukt mye, til tross for restriksjonene som er karakteristiske for det. CIS.-1,2-cykloheksanediol ble først mottatt av v.v. Markovnikov i 1878 av hydroksylering av cykloheksen med vandig oppløsning av kaliumpermanganat ved 0 0 C.

Denne metoden i fremtiden ble utviklet i verkene til den russiske forskeren e.e. Wagner, derfor syn- Hydroksilisering av alkener under virkningen av vandig løsning av permanganatkalium kalles Wagner-reaksjonen. Kaliumpermanganat er et sterkt oksidasjonsmiddel som ikke bare kan hydroksy dobbeltbindingen, men også splittet den formende vicinal diol. For å unngå ytterligere spaltning av glykoler, er det nødvendig å nøye overvåke reaksjonsbetingelsene. Utgangene av glykoler samtidig er vanligvis små (30-60%). De beste resultatene oppnås i hydroksylering av alkener i et svakt alkalisk medium (pH ~ 8 9) ved 0-50 med en fortynnet med 1% vandig løsning av KMNO 4.


I utgangspunktet, under oksydasjonen av alkenes permanganatkalium, dannes en syklisk eter av manganyre, som umiddelbart hydrolyseres til den vicinal diola.

Den cykliske esteren av manganyre som et mellomprodukt ble ikke isolert, men formasjonen følger av eksperimenter med merket 18 på kaliumpermanganat: Både oksygenatomer i glykol er merket når alkina er oksidasjon KMN 18 o 4. Dette betyr at både oksygenatomer beveger seg fra oksydanten, og ikke fra løsningsmiddelvannet, som er i god i henhold til den foreslåtte mekanismen.
En annen metode syn- Hydroksilisering av alkener under virkningen av OSMIA oksyd (VIII) OSO 4 ble foreslått av R. Kryga i 1936. Osmia Tetraoksid er en fargeløs, flygende, krystallinsk, godt løselig på luft, dioksan, pyridin og andre organiske løsningsmidler. Når osmiumtraoksydet samhandler med alkaner i luften eller dioksanen, dannes et svart utfelling av osmuminsyre-sykliskester - OSMAT, som lett kan isoleres individuelt. Tilsetningen av OSO 4 til dobbeltbindinger er merkbart akselerert i løsningen i pyridin. Nedbrytningen av osmastene til de vicinale glykolene oppnås ved virkningen av vandig natriumhydrosulfitt eller hydrogensulfid.

Produktutganger syn-Hyndroxylering av alkener i denne metoden er betydelig høyere enn ved bruk av permanganat som et oksidasjonsmiddel. En viktig fordel ved Krig-metoden er fraværet av oksidative splittingsprodukter av alkener som er karakteristisk for permanganatoksydasjon.


Osmia Tetraoksid er et veldig dyrt og vanskelig å nå reagens, og det er giftig. Derfor brukes Osmia Oxide (VIII) i syntesen av små mengder vanskelige stoffer for å oppnå den høyeste utgang fra diolen. For å forenkle syn-Hyndroksylering av alkener under virkningen av OSO 4 ble utviklet av en teknikk som tillater oss å bruke bare de katalytiske mengder av dette reagenset. Alkenhydroksylering utføres ved anvendelse av hydrogenperoksid i nærvær av OSO 4, for eksempel:

Som konklusjon av denne delen gir vi stereokjemiske forhold mellom alken cIS.- eller transe- Konfigurasjon og konfigurasjon av den dannede vicinal diol, som kan være cIS.- eller transe-Zeter, erytro- eller treo.-Mat meso.- eller D, L.-Form, avhengig av varamedlemmer i Alken:

Lignende stereokjemiske relasjoner observeres i andre reaksjoner syn- eller anti.- under flertallet av tilførsel av hydrogen, halogenerater, vann, halogen, borhydrider og andre reagenser.
Kompilering av ligninger av redoksreaksjoner som involverer organiske stoffer
I Kommunikasjon med introduksjonen som den eneste form for den endelige sertifiseringen av videregående opplæring av en enkelt statseksamen (bruk) og overgangen av videregående skole for profilopplæring blir stadig mer relevant for utarbeidelsen av videregående studenter for å oppfylle de mest "dyre "Oppgaver av del" med "test av eksamen i kjemi. Til tross for at de fem oppgavene i delen "C" regnes som forskjellige: de kjemiske egenskapene til uorganiske stoffer, kjederne for omdannelsen av organiske forbindelser, de beregnede oppgavene - alle er på en eller annen måte er tilknyttet nettopp med Redox reaksjoner (OSR). Hvis hovedkunnskapen til teorien om OSR læres, kan du korrekt utføre de første og andre oppgavene helt, og den tredje er delvis. Etter vår mening er en betydelig del av suksess når du utfører en del "C" nettopp i dette. Erfaring viser at hvis studentene studerer uorganisk kjemi, blir studentene godt håndtert med oppgavene for å skrive OSR-ligningene, forårsaker de lignende oppgavene for organisk kjemi dem store vanskeligheter. Derfor, gjennom hele studien av hele løpet av organisk kjemi i profilklasser, prøver vi å danne ferdighetene til samlingen av OPR-ligningene i videregående studenter.
Når vi studerer de komparative egenskapene til uorganiske og organiske forbindelser, introduserer vi studenter som bruker graden av oksidasjon (C.O.) (i organisk kjemi, først og fremst karbon) og metoder for bestemmelse:
1) Beregning av medium S.O. karbon i det organiske materialet molekylet;
2) Definisjon av S.O. Hvert karbonatom.
Vi angir i hvilke tilfeller det er bedre å bruke en eller annen måte.
Artikkelen ble publisert med støtte fra selskapet "Geo-Engineering", som representerer produktene i markedet under merkevaren "Profkresla". Omfanget av selskapet - produksjon, salg og installasjon av stoler og stoler for ulike haller. Den høye profesjonaliteten til de ansatte og deres egne produksjonsanlegg gir deg raskt og effektivt å gjennomføre prosjekter av enhver grad av kompleksitet. Alle produkter under merkevaren "Profkresla", være en tonatral stoler, seter for venterom eller stoler for utdanningsinstitusjoner, skiller moderne og ergonomisk design, samt høy slitestyrke, styrke og komfort. Fra det enorme spekteret av produkter som presenteres i katalogen på nettstedet Profkresla.ru, kan du alltid velge modellene som er best relevante for bedriftens stil som er vedtatt i din bedrift. Hvis du fortsatt har problemer med valget, er selskapets eksperter alltid klare til å gi råd, bidra til å bestemme modellen, hvoretter det er å forberede prosjektet på stedet for å produsere alle nødvendige målinger og installasjon.
S Studien av emnet "Alkana" viser at prosessene for oksidasjon, forbrenning, halogenering, nitrasjon, dehydrering, dekomponering relaterer seg til redoks prosesser. Når du skriver ligningene i forbrenningsreaksjoner og dekomponering av organiske stoffer, er det bedre å bruke gjennomsnittsverdien av C.O. Karbon. For eksempel:
Vi trekker oppmerksomhet til første halvdel av den elektroniske balansen: på karbonatomet i brøkdelen av S.O. Nettet er 4, slik at beregningen av elektronoverføring utføres i henhold til denne koeffisienten.
I andre tilfeller, når vi studerer emnet "Alkans", bestemmer vi verdiene til S.O. Hvert karbonatom i forbindelsen, tegner oppmerksomheten til elevene til sekvensen av erstatning av hydrogenatomer i primære, sekundære, tertiære karbonatomer:


Således oppsummerer vi studenter i den konklusjon at prosessen med substitusjon i tertiær, deretter i sekundær, og siste gang - i primære karbonatomer.
S Studien av emnet "Alkenes" anser oksidasjonsprosesser avhengig av strukturen av alkenet og reaksjonsmediet.
Når alkenene oksyderes av den konsentrerte oppløsning av kaliumpermanganat KMNO 4 i et surt medium (stiv oksidasjon), oppstår en pause - og -konger med dannelsen av karboksylsyrer, ketoner og karbonoksid (IV). Denne reaksjonen brukes til å bestemme posisjonen til dobbeltbindingen.
Hvis dobbeltbindingen er på enden av molekylet (for eksempel ved Bouthena-1), er en av oksidasjonsproduktene myresyre, lett oksyderes til karbondioksid og vann:

Vi understreker at hvis i Alkin-molekylet omfatter karbonatomet ved et dobbeltbinding to karbonstituenter (for eksempel i et 2-metylbuten-2-molekyl), så når det er oksidasjon, oppstår ketondannelsen, fordi konverteringen av et slikt atom i et atom i karboksylgruppen er umulig uten å bryte C-C-kommunikasjon, relativt stabil under disse forholdene:

Vi angir at hvis Alkinmolekylet er symmetrisk og dobbeltbindingen er inneholdt i midten av molekylet, dannes bare en syre under oksidasjon:

Vi informerer om at funksjonen ved oksydasjonen av alkener der karbonatomer ved et dobbeltbinding omfatter to karbonradikaler, er dannelsen av to ketoner:


Tatt i betraktning oksydasjon av alkener i nøytral eller svakt alkaliske medier, fokuserer oppmerksomheten til videregående studenter på det faktum at oksydasjonen i slike forhold er ledsaget av dannelsen av dioler (ductomicalkoholer), og hydroksylgruppene er forbundet med karbonatomer , mellom hvilke det var en dobbeltbinding:

I På samme måte vurderer vi oksydasjonen av acetylen og dets homologer, avhengig av hvilket miljøet fortsetter. Så, vi spesifiserer at oksidasjonsprosessen i et surt miljø er ledsaget av dannelsen av karboksylsyrer:

Reaksjonen brukes til å bestemme strukturen av alkiner på oksidasjonsprodukter:

I nøytral og lav-alkalisk media er oksydasjonen av acetylen ledsaget av dannelsen av passende oksalat (salter av oksalsyre), og oksydasjonen av homologer er en nedbrytning av trippelbindinger og dannelsen av karboksylsyresalter:

I CE-reglene utføres med studenter på spesifikke eksempler, noe som fører til en bedre absorpsjon av teoretisk materiale. Derfor, når du studerer oksydasjonen av arenaen i ulike miljøer, kan studentene selvstendig foreslå at syreformasjon bør forventes i et surt medium, og i alkaliske salter. Læreren vil bare bli avklart hvilke reaksjonsprodukter som er dannet, avhengig av strukturen til den relevante arenaen.
Viser på eksemplene som gomologer av benzen med en sidekjede (uavhengig av lengden) oksyderes av et sterkt oksidasjonsmiddel til benzosyre med karbonatom. Gomezol homologer oksyderes av kaliumpermanganat i et nøytralt medium for å danne kaliumsalter av aromatiske syrer.
5c 6 h 5 -CH3 + 6KMNO 4 + 9H2SO4 \u003d 5c 6 H5COOH + 6MNSO4 + 3K2SO4 + 14H20,
5c 6 h 5-C2H5 + 12kmno 4 + 18H2SO4 \u003d 5C 6H5OOH + 5CO2 + 12MNS04 + 6K2SO4 + 28H20,
C 6 H 5 -CH3 + 2KMNO 4 \u003d C 6 H 5 Kok + 2MNO 2 + KOH + H 2 O.
Vi understreker at hvis det er flere sidekjeder i molekylet, så i et surt medium oksyderes hver av dem i henhold til et karbonatom til karboksylgruppen, som et resultat av hvilke polybentaromatiske syrer dannes:

S Olliniserte ferdigheter for å kompilere OSR-ligningene for hydrokarboner lar deg bruke dem når de studerer seksjonen "oksygenholdige forbindelser".
Når man studerer temaet "Alkohol", utgjør studentene uavhengig likning av alkoholoksydasjon ved hjelp av følgende regler:
1) Primæralkoholer oksyderes til aldehyder
3CH3 -CH2OH + K 2 CR 2O7 + 4H2SO4 \u003d 3CHO3 -CHO + K2SO4 + CR2 (SO 4) 3 + 7H20;
2) Sekundære alkoholer oksyderes til ketoner

3) For tertiære alkoholer er oksidasjonsreaksjonen ikke karakteristisk.
For å forberede eksamenen gir læreren tilrådelig ytterligere opplysninger til de angitte egenskapene, som utvilsomt vil være nyttig for studenter.
Med oksydasjonen av metanol med en surgifisert oppløsning av kaliumromanganat eller kaliumdikromat, dannes CO2, primære alkoholer under oksidasjon, avhengig av betingelsene for reaksjonen, kan danne ikke bare aldehyder, men også syrer. For eksempel, oksydasjonen av etanol med dikromatkalium på kalde ender med et eddiksyrebelt, og når oppvarmet - acetaldehyd:
3CH3 -CH2OH + 2K 2 CR 2O7 + 8H2SO4 \u003d 3CH3 -COOH + 2K2SO4 + 2CR2 (SO 4) 3 + 11H20,
3CH3 -CH2OH + K 2 CR 2O7 + 4H2SO4 3CHO 3 -CHO + K2SO4 + CR2 (SO 4) 3 + 7H20.
Vi husker igjen studenten på effekten av mediet på produktene av alkoholoksidasjonsreaksjoner, nemlig: den varme nøytrale oppløsningen av KMNO 4 oksiderer metanol til kaliumkarbonat og de gjenværende alkoholer - til salter av de tilsvarende karboksylsyrer:

Når du studerer emnet "aldehyder og ketoner", legger du vekt på oppmerksomheten til elevene på det faktum at aldehyder er lettere enn alkoholer, oksideres i de passende karboksylsyrer, ikke bare under virkningen av sterke oksidasjonsmidler (oksygen, surgjorte løsninger av KMNO 4 og K 2 Cr 2 O 7), men under påvirkning av svak (ammoniakkløsning av sølvoksyd eller kobberhydroksyd (II)):
5CH3 -CHO + 2KMNO 4 + 3H2SO4 \u003d 5CH3 -COOH + 2MNSO 4 + K2SO4 + 3H20,
3CH3 -CHO + K 2 CR 2O7 + 4H2SO4 \u003d 3CH3 -COOH + CR 2 (SO 4) 3 + K2SO4 + 4H20,
CH3 -CHO + 2OHCH3-COONH 4 + 2AG + 3NH3 + H 2 O.
Spesiell oppmerksomhet er betalt til oksydasjonen av metanal med ammoniakkløsning av sølvoksid, siden I dette tilfellet dannes ammoniumkarbonat, og ikke myresyre:
Hchex + 4OH \u003d (NH4) 2 CO 3 +4AG + 6NH3 + 2H20.
Som våre mange års erfaring viser, øker den foreslåtte metoden for å lære videregående skoleelever å kompilere ORP-ligningene som involverer organiske stoffer sitt endelige resultat av eksamen i kjemi i flere punkter.
Oksydasjonen av alkener permanganatkalium i et alkalisk medium under oppvarming (stive forhold) fører til ødeleggelsen av deres karbonskjelett på dobbeltforbindelsesstedet. På samme tid, avhengig av antall alkylgrupper som er forbundet med vinylfragmentet, kan to karboksylsyrer, syre og keton eller to ketoner oppnås:
UPR.11.Hvilket produkt dannes under oksydasjonen av cykloheksien (A) fortynnet kaliumpermanganatoppløsning i kulde og (b) konsentrert kaliumpermanganatløsning med etterfølgende forsuring.
UPR.12.Hvilke produkter dannes fra 1,2-dimetylcykloheksen ved sin (a) katalytisk hydrogenering, (b) oksydasjon av kalium permanent kaliumpermanganat i kulde, (c) ozonering med etterfølgende reduksjon av splitting.
6.5. Etylenoksydasjon i acetaldehyd
Oksydasjonen av etylenluftseksygen i nærvær av palladiumklorider (II) og kobber (II) fører til dannelsen av acetaldehyd ( Wheaker prosess):
 (63)
(63)
etanal (Acetaldehyd)
6.6. Klorering etylen
Vinylklorid oppnås ved klorogen etylen:
6.7. Oksidativ ammonolyse
Oksydasjonen av hydrokarboner med luft oksygen i nærvær av ammoniakk fører til transformasjonen av metylgruppen i cyano-gruppen. Slike oksydasjon kalles oksidativ ammonolyse. Akrylonitril oppnås ved oksydasjon av propylen.
akrylonitril
Den oksidative ammonolysen av metan oppnås ved en sinylsyre:
 (66)
(66)
7. Hydraulering av alkener (oksosyntese)
Ved temperaturer fra 30 til 250 o C og et trykk på 100-400 atm. I nærvær av dicobaltoktakarbonyl, inngår alkener hydrogen og karbonmonoksid med dannelsen av aldehyder. Blandingen av isomerer oppnås vanligvis:
Mekanisme:
1. Dekorasjon av Liganda
2. Vedlegg Etylen

3. Innføringen av etylen

4. Feste Liganda.
5. Implementering av CO
6. Oksidativ hydrogenvedlegg

7. Recovery Pickup

8. Tilgang til karbennes og karbenoider
De siste årene er det lagt stor vekt på organisk kjemi til forbindelsene med bivalent karbon - karbenas. De fleste av karbentene er ustabile og umiddelbart etter at dannelsen reagerer med andre tilkoblinger.
8.1. Karbenovs struktur
Unnoticed Carben: CH2, også kalt metylen, kan være i en singlet eller triplettform. I singletformen av karben er to ukonvisende elektroner med parede spinn plassert på samme orbitale, mens i triplettform er to uberørte elektroner med parallelle spinn plassert på to orbitaler av samme energi. Ulike elektroniske konfigurasjoner av singlet og triplettkarbenes reflekteres både i forskjellig geometri av disse partiklene og i ulike kjemiske aktiviteter. Et bivalent karbon karbonatom er plassert i en SP 2-hybrid-tilstand, begge elektroner er plassert på SP 2-flexidal orbitaler (bølge), og en p-orbital (NSMO) er fri. Triplet Carben er preget av sp-hybridisering av bivalent karbon; Samtidig er to uberørte elektroner plassert på to p-orbitals, dvs. triplet Carben er en biradisk. Vinkelen til H-H-H for singlet metylen, i henhold til spektraldata, er 102-105 0, og for triplet metylen øker denne vinkelen til 135140 o. Dette tilsvarer en høyere stabilitet av triplet-metylen. I henhold til kvantemekaniske beregninger er tripletmetylen virkelig 10 kcal / mol mer stabil singlet metylen.

Substituentene forårsaker imidlertid en endring i den relative stabiliteten til disse to former for karben. For Dialkyl Carben er et triplettskjema også mer stabilt singlet, men for Digalokarben : Chal 2, og andre karbnes med substituenter som inneholder et vannet par elektroner, er hovedstaten en singlet. Valensvinkelen til C1-C-C1 for Dichlorkarbena, som er lik 106 O, er godt i samsvar med singletformen. Høyere stabilitet av singletformen til digalokarbens sammenlignet med triplet, tilsynelatende på grunn av stabiliseringen på grunn av dampparet av elektroner heteroatom

Slike stabilisering av trippelformen av Digokarben er umulig. I henhold til dataene i kvantemekanisk beregning, er energien til singlet - den triplettovergangen for Dichlorkarben 13,5 kcal / mol.
A. Dichlorkarben
For å generere Digalokarbenov utvikles metoder basert på reaksjonen av elmins av halogenhydrogen fra trigalo-gener under virkningen av sterke grunner. Denne metoden var historisk den første, med hjelp av hvilken den første av Karbenov ble generert som en mellomliggende - Dichlorkarben (J. Hein 1950). Ved interaksjon med alvorlige baser av kloroform (RKA kloroform er ~ 16), er bromormal (RCA \u003d 9) og andre trigogomenomet dannet en anion som stabiliseres ved å eliminere halogenidionen med dannelsen av digokarben. Handling på kloroform av sterke baser er oppnådd av Dichlorkarben:
dichlorkarben
Som en base kan litiumorganiske forbindelser i det likegyldige aprotiske medium også benyttes. Deretter kan under -100 0 C festes i dannelsen av triklormetyl litium som et mellomprodukt.

Ved hjelp av sterke baser som RLI, kan du generere slått fra 1,1-tigningenes derivater

I de siste årene, for å generere Digalokarben i stedet n.-Butilitia er mye brukt som en base av bis (trimetylsilyl) natriumamid.
Samtidig er kjemisk inert kjærlig [bis (trimethylsilyl) amid] preget. BIS (trimetylsilyl) natriumamid, i motsetning til n-butyl litium, kan separeres i en inert atmosfære i en tørr form. I praksis kan de essensielle løsningene brukes oftere, som kan lagres ved romtemperatur i lang tid.
Dichlorkarben kan også genereres med termisk decarboxylering av natriumtørrkloracetat:
En av de mest rimelige moderne metodene for å generere Dichlorkarbena fra kloroform under virkningen av natriumhydroksyd i forholdene for grensesnittkatalyse vil bli beskrevet i detalj senere.
Dichlorkarben slutter seg til alken, som gir diklorcyklopropans. Tilkoblingen skjer Stereospesifikk - Konfigurasjonen av det opprinnelige alkenen er bevart i reaksjonsproduktet - Cyclopropan:
 (69)
(69)
transe-2-Buten transe-1,2-dimetyl-3,3-
diklorcyklopropan
 (70)
(70)
cIS.-2-Buten q.fra-1,2-dimetyl-3,3-
diklorcyklopropan
 (71)
(71)
7,7-diklorubicykloheptan
Ved gjenoppretting av 1,1-digalocyklopropans under virkningen av litium i mpem.-Butylalkohol, sink i eddiksyre eller natrium i flytende ammoniakk, er begge halogenatomer erstattet av hydrogen. Dette er en av de generelle metodene for å produsere syklopropanderivater.
bicyclicheptan.
UPR. elleve.Fullfør reaksjonen:


(Z) -3-metyl-2-penten metylenecykloheksan
Svar

B. Methylen
Methylen kan oppnås ved dekomponering av diazometan. Diazometan er et relativt ustabilt stoff som dekomponerer ved bestråling på nitrogen og metylen.
 (73)
(73)
diazometan
Methylen: CH2 med et diazometanfotolidium dannes i en mindre stabil singletform. Singlet metylen Under reaksjonsbetingelsene som følge av kollisjoner med diazometan eller nitrogenmolekyler, mister det raskt energi og blir til en mer stabil triplet-metylen.
For singletkarbenis er det preget av synkron tilkobling til dobbeltbindingen av alken med den fullstendige bevaring av geometrien ved en dobbeltbinding (reaksjonscykloperforbindelse). Vedlegget av singletformen på karbenet for en dobbeltbinding oppstår, og dermed strengt stereospesifikke.
B. Reaksjonssimoner-Smith.
Effektiv og eksperimentelt, er det svært enkel metode for å konvertere alkener i cyklopropanderivater basert på reaksjonen av alkener med Iodistan-metylen og sink og kobberlegering. Denne reaksjonen ble åpnet i 1958 av Simmons og Smith og vant umiddelbart stor popularitet i syntesen av cyklopropanderivater. Aktiv partikkel i denne reaksjonen er ikke karben : CH2, og karbenoid-jodid-jodetylcykling iznch 2 I, dannet av samspillet mellom metylenjodid og sinkkobberpar.
diodmethan iodomeomethylcinodid.
(Ekte Simmons Smith)
 (75)
(75)
Reaksjonen passerer gjennom følgende mekanisme:

Simmons Smith-reaksjonen er en veldig praktisk metode for å konvertere alkener i cyklopropan.
UPR. 12.Fullfør reaksjonen:


Svar
 (76)
(76)
metylenecyklopentan spiroheptan.
 (77)
(77)
styren syklopropylbenzen.